Best Open-Source Software for Molecular Interaction Analysis
Molecular interaction analysis forms the foundation of modern drug discovery, enabling researchers to understand how molecules bind to biological targets and predict therapeutic activity. Open3DQSAR stands among the leading open-source platforms for high-throughput chemometric analysis of molecular interaction fields. This software processes complex three-dimensional molecular data, builds predictive models, and provides insights crucial for pharmaceutical research. Its open-source nature ensures transparency, reproducibility, and accessibility for research institutions worldwide without prohibitive licensing costs.
Core Capabilities of Molecular Analysis Software
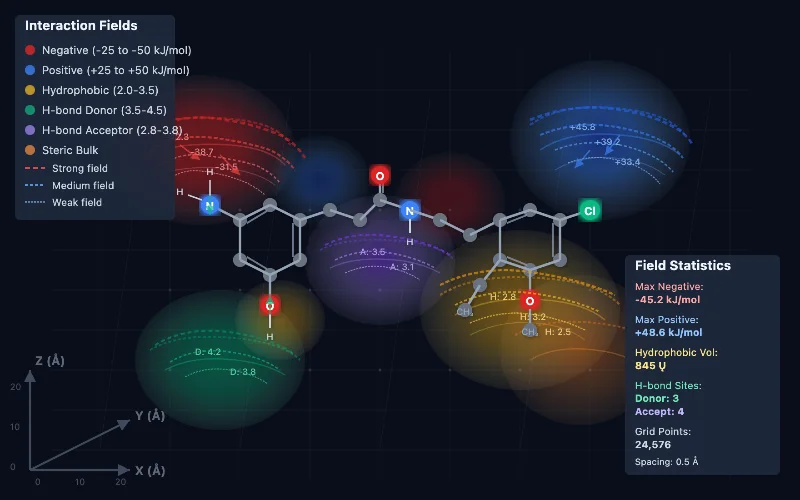
Molecular interaction analysis software processes spatial data representing how molecules interact with their environment. These tools generate molecular interaction fields encoding electrostatic potential, hydrophobic regions, and hydrogen bonding sites. Advanced chemometric techniques then analyze these fields to identify patterns correlating with biological activity.
- Three-dimensional field generation creates spatial representations of molecular properties across grid points
- Partial Least Squares regression builds predictive models correlating molecular features with biological activity
- Variable selection algorithms identify the most informative molecular regions for activity prediction
- Cross-validation procedures ensure model reliability and prevent overfitting to training data
Software Platform Comparison
Various platforms offer molecular interaction analysis capabilities with different strengths and licensing models.
| Platform | Primary Focus | License Type |
|---|---|---|
| Open3DQSAR | Chemometric MIF analysis | Open Source |
| GRID | Field calculation | Commercial |
| CoMFA | Comparative analysis | Commercial |
| AutoDock | Molecular docking | Open Source |
"Open-source molecular analysis tools democratize access to sophisticated computational methods, enabling researchers worldwide to perform rigorous drug discovery studies regardless of institutional budget constraints."
Applications in Drug Development
Molecular interaction analysis guides multiple stages of drug discovery. Early-stage research uses these tools to identify promising molecular scaffolds, while lead optimization employs them to refine compound properties for improved efficacy and reduced side effects. The ability to predict activity before synthesis saves considerable time and resources by focusing experimental efforts on the most promising candidates. Open3DQSAR's integration with other molecular modeling software creates comprehensive workflows spanning from initial virtual screening through detailed binding analysis, supporting the entire computational drug discovery pipeline with transparent, verifiable methodologies.
